The FDA’s 510(k) pathway clears most Class II medical devices for the U.S. market — but it’s a high-stakes process where nearly one-third of submissions are rejected before review. Avoidable errors like incomplete documentation or misaligned testing can delay launches for months. Strong, centralized document control can mean the difference between a smooth clearance and a costly setback.
For most medium-risk (Class II) medical devices, the US Food and Drug Administration’s (FDA) 510(k) submission process is the primary pathway to market. No less than 99% of the >155,000 devices approved or cleared by the FDA since 1976 used the 510(k) pathway. Yet, it’s far from a rubber stamp: almost a third of submissions were rejected before they even reached substantive review in 2022. And more than two-thirds of FDA 510(k) submissions result in requests for additional Information
The stakes are high — a single oversight can lead to an early rejection adding months of delay, derailing launch timelines, and eroding your competitive advantage in introducing your product to the American market. They are often caused by incomplete documentation, misaligned testing evidence, or quality system gaps.
A new-generation Document Management System can help MedTech companies reduce these risks by centralizing documentation, enforcing quality and compliance workflows, and providing full traceability throughout the submission process.
Understanding the 510(k) Process
To receive clearance, manufacturers must demonstrate that their device is substantially equivalent to an already-cleared “predicate” device in terms of safety and effectiveness. This requires detailed technical, safety, and performance data, along with proof of compliance with FDA-recognized quality standards.
There are three main 510(k) types:
- Traditional 510(k): The standard pathway, supported by full documentation and testing.
- Special 510(k): For device modifications that do not alter intended use or fundamental technology.
- Abbreviated 510(k): Leveraging recognized standards and guidance documents to streamline review.
While the submission format is standardized, the supporting evidence is unique to each device, meaning document control and traceability are critical.
Common Pitfalls in 510(k) Submissions
FDA guidance and feedback show that many rejections are avoidable. Common issues include:
- Incomplete or inconsistent documentation between submission sections.
- Quality system documentation gaps during premarket review.
- Unverified or outdated test results that fail to meet FDA-recognized methods.
- Lack of linkage between design history, risk management, and performance data.
These issues often stem from fragmented document management systems, siloed teams, and manual review processes.
Building a Stronger 510(k) Submission
MedTech manufacturers can improve their chances of a successful submission with a centralized, compliant, and collaborative platform to manage every document and approval in the 510(k) process.
Key capabilities might include:
- Centralizing information storage and processing so all teams work from the same, current documentation.
- Maintaining full traceability by linking predicate devices’ test results, risk assessments, and design history files.
- Using structured review processes to catch inconsistencies well before submission.
- Keeping quality system records audit-ready at all times by maintaining a complete version history with time-stamped approvals to support FDA inspections or audits.
- Collaborating effectively without compromising on security, safety and access controls: Enable internal teams and external partners to contribute while preserving document integrity and access controls.
Beyond satisfying regulatory requirements, these practices reduce rework, speed up internal approvals, and shorten time to market.
Tools That Support Quality and Compliance
For organizations seeking to strengthen document control, ensure manufacturing excellence, and maintain regulatory compliance, modern document management technology can help consolidate workflows, reduce manual effort, and ensure records are complete and up to date.
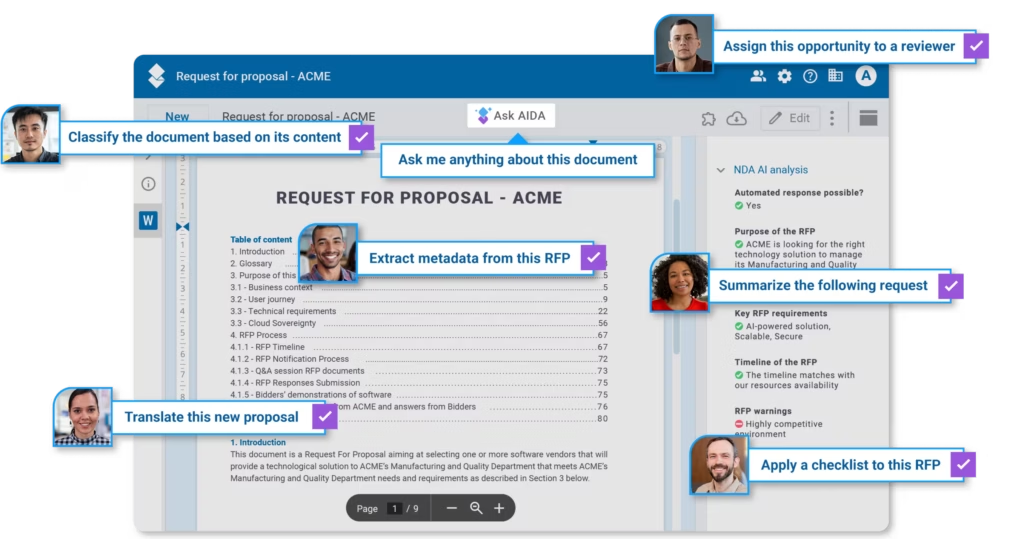
AODocs, for example, offers an AI-powered quality management system that unifies manufacturing, safety, and compliance records in a cloud-native environment. This type of centralized, traceable system can play a key role in supporting complete, consistent, and audit-ready submissions — ultimately increasing the likelihood of a smooth 510(k) clearance process.
Learn More:
Watch: How AODocs’ AI-Powered QMS Works
Explore AODocs Solutions for Quality Management